
化学構造を手掛かりにしたデータサイエンスの手法で、
天然物化合物の生合成経路の予測に成功!
有機分子の特性や薬剤設計の研究への活用期待
【概要】
奈良先端科学技術大学院大学(学長:横矢直和)先端科学技術研究科 データ駆動型サイエンス創造センター、情報科学領域 計算システムズ生物学研究室(兼務)の小野直亮准教授のグループは、AI(人工知能)技術として普及している深層学習の応用の一つである「グラフコンボリューション(畳み込み)ニューラルネットワーク法」を活用し、生物が有する多様な天然物化合物が合成される起点となる物質を予測するモデルの構築に成功しました。
この研究は、生命情報学における国際論文誌BMC Bioinformatics に採録されました。昨今、画像 認識の分類などで広く活用されている深層学習の手法である畳み込みニューラルネットワークを、天然物化合物のように多数の原子が結合した複雑なネットワーク構造に応用することで、分子の合成経路 の予測といった細胞内の複雑な現象を学習する方法として利用し、高精度な予測が可能になりました。
多種多様な分子から新しい機能を持つ分子を探し出すケモインフォマティクス(化学情報科学)の 分野をはじめ、ビッグデータサイエンス、データ駆動型サイエンス、さらにはビッグデータバイオロジ ーなどの分野の複雑なデータに応用していくことで、今後さまざまな発見を導く可能性が開けたと考えています。
【掲載論文】
Ryohei Eguchi, Naoaki Ono (責任著者), Aki Hirai (Morita), Tetsuo Katuragi, Satoshi Nakamura,Ming Huang, Md. Altaf-Ul-Amin, Shigehiko Kanaya,
Classification of alkaloids according to the starting substances of their biosynthetic pathwaysusing graph convolutional neural networks,
BMC Bioinformatics (2019)
【背景と目的】
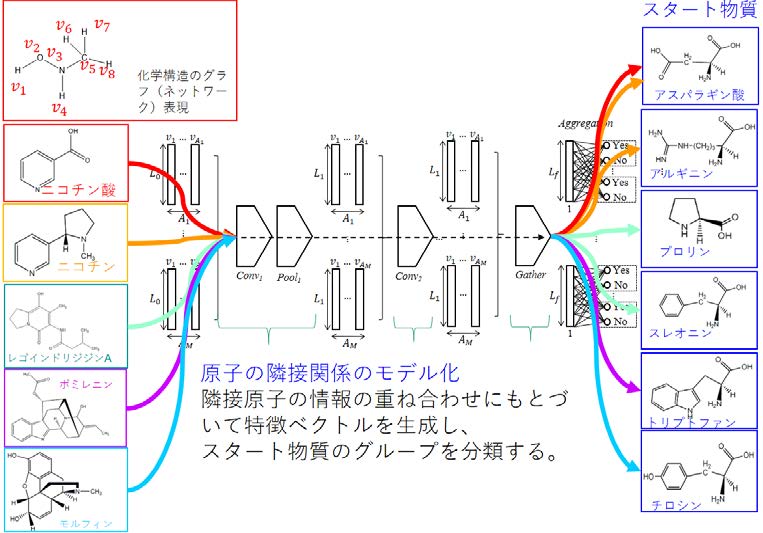
顔認識や手書き文字認識など、画像データを自動的に分類する問題では、近年画像データの一部からパターンを抽出する計算(畳み込みフィルター)を何段階も積み重ね、全体の画像から効率よく特徴量を求めるコンボリューション(畳み込み)ネットワーク(CNN)をはじめとした深層学習の手法の発展により、その分類精度は飛躍的に向上してきました。この技術を分子構造の表現に応用することにより、これまで原子の結合関係などから分子構造の情報を機械的に定量化していた手法をより効率よく最適化できるようにしたものが分子グラフコンボリューションニューラルネットワーク(MGCNN)法です。このほど、本学データ駆動型サイエンス創造センターの小野直亮准教授のグループが、このMGCNN 法を用い、生物において生合成される天然物を、合成の起点となる分子、スタート物質に基づいて高精度で分類する方法を世界に先駆けて開発した成果が、BMC Bioinformatics にアクセプトされました。
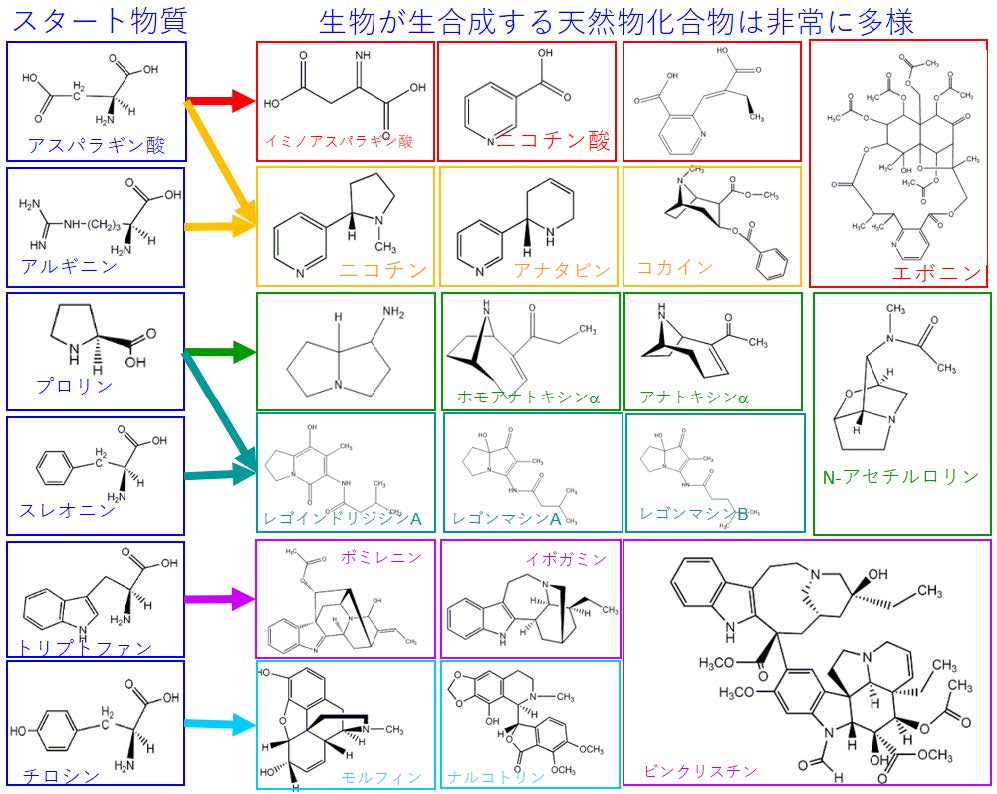
地球上の様々な生物により、あわせて約100 万種にのぼる天然物化合物が合成されていることが知られています。その中で、窒素を含む代謝物(アルカロイド類)は、それぞれ合成の起点となるアミノ酸が決まっていると考えられています(図1)。しかしながら現状では、2 万種類近いアルカロイド類のうち、1 千種にも満たない一部の分子についてしか具体的な起点となる物質の種類はわかっていません。
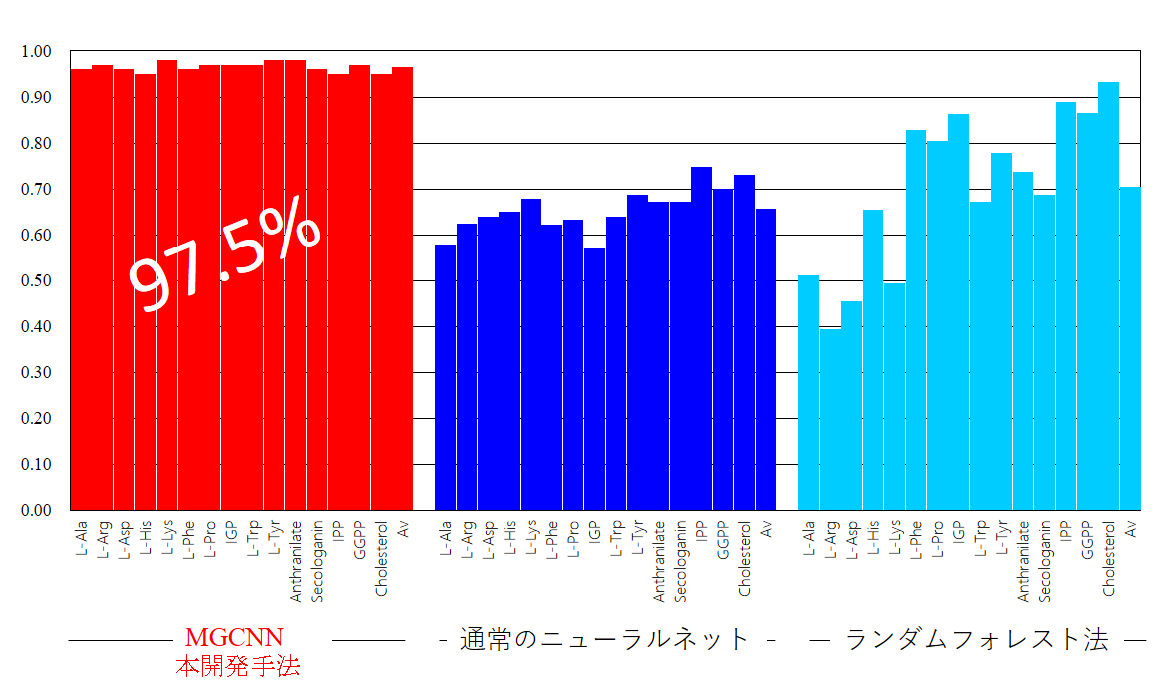
こうしたことから、まずスタート物質がわかっている分子を教師データとし、MGCNN 法を用いて学習させることにより、それぞれのアルカロイド分子のスタート物質を予測するためのモデルを開発しました(図2)。本方法の分類精度は97.5%とこれまでの機械学習と比べても格段と高く(図3)、これまでスタート物質が不明だった代謝物についても、その合成の起点を高精度で予測できると期待されます。
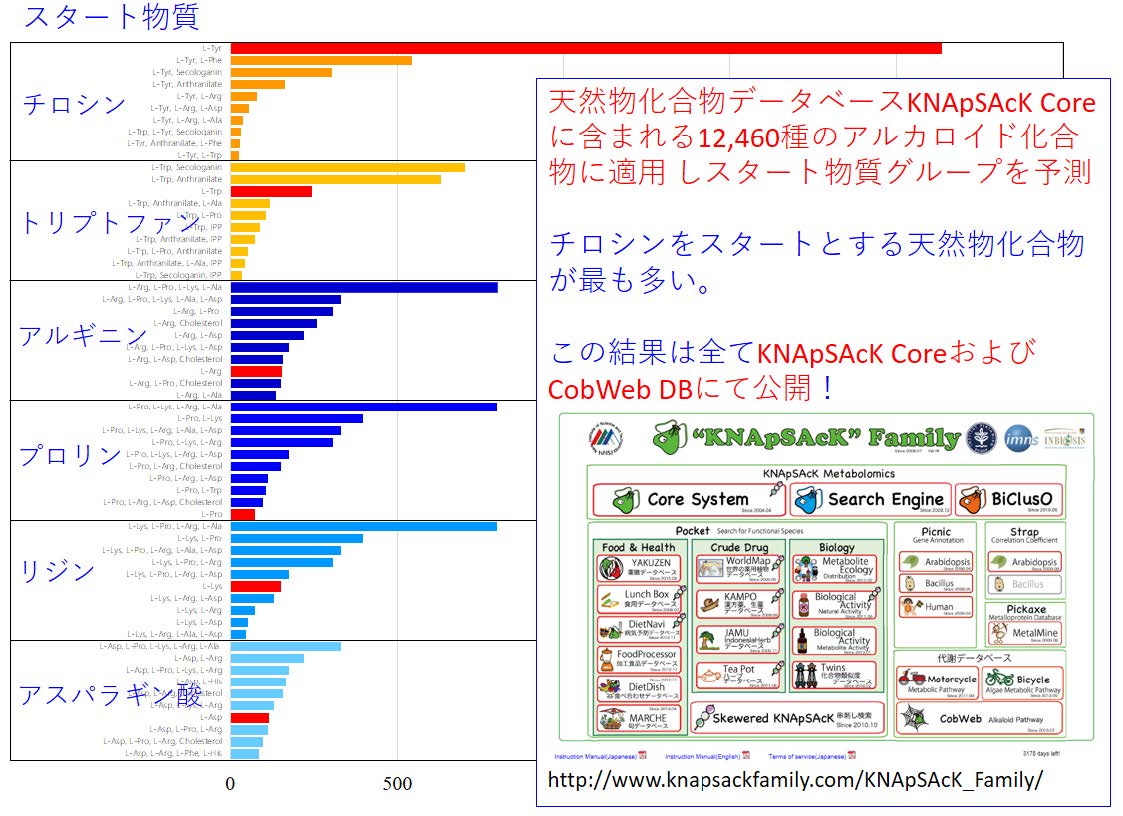
そこで、この手法を用いて本研究室で開発を進めている二次代謝産物データベース「KNApSAcKデータベース」に登録されている天然物化合物約50,000 種のうち12,460 種にのぼるアルカロイド代謝物全てについてそのスタート物質を予測したところ、アミノ酸の一種であるチロシンが最も多く使われている(図4)など、特徴的な分布が見られるという結果が得られました。これらのデータは、研究者ならびに本分野に興味のある方すべてが活用できるよう、全て私たちのWeb サイト(http://www.knapsackfamily.com/KNApSAcK_Family/) で公開しています。
【今後の展開】
MGCNNは、化学構造に基づく機械学習に広く応用可能な手法であり、有機分子の分類のみならず、化学的な物性の定量や、たんぱく質との相互作用の予測など、様々な問題に利用できます。本研究で示した結果は、例えばドラッグデザインなどの分野での活用が期待できると言えます。この手法の応用は、化学構造に基づく予測が必要な、ケモインフォマティクス、マテリアルインフォマティクス(物質材料情報科学)、さらにはバイオインフォマティクス(生命情報科学)において、分子の特性を予測し、期待する構造をデザインするための基盤技術として、今後の広く活用されていくことになると考えています。
【本研究内容についてコメント出来る方】
奈良先端科学技術大学院大学 先端科学技術研究科 ソフトウエア工学研究室
教授 松本 健一
TEL:0743-72-5310 E-mail:matumoto@is.naist.jp
【本プレスリリースに関するお問い合わせ先】
奈良先端科学技術大学院大学 データサイエンス創造センター
准教授 小野 直亮
TEL:0743-72-5952 E-mail:nono@is.naist.jp
奈良先端科学技術大学院大学 先端科学研究科 情報科学領域 計算システムズ生物学研究室
教授 金谷 重彦
TEL:0743-72-5952 E-mail:skanaya@gtc.naist.jp